LoG conference, ‘22,
LoG conference Day 4 Youtube

Lecturer: Djork-Arné Clevert
- 2022.05.19~09.22: Bayer Pharma, Head of Machine Learning Research (All work presented today were published during Bayer.)
- 2022.10.22~: Pfizer, Head of Machine Learning Research
Summary
- Most ML methods are focused on early drug discovery part.
- Major applications of ML in drug discovery include:
- ADMET modeling
- Representation learning
- Conditional de novo hit design
- Inverse molecule modeling
Drug Discovery vs Drug Development
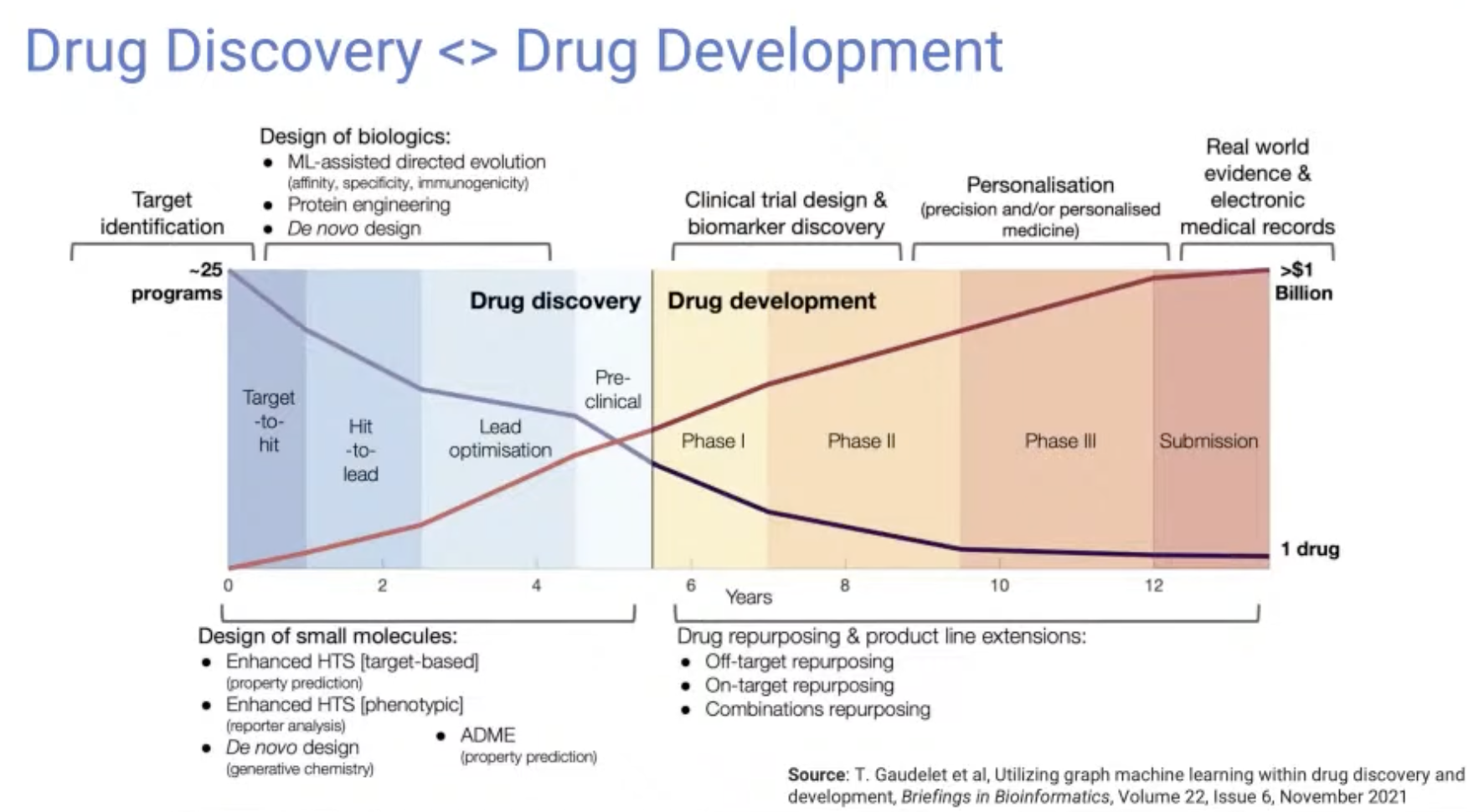
- Drug discovery: the early part (Hit identification ~ Pre-clinical phase)
- Drug development: the later part (Clinical trials phase 1 ~ Phase 4)
- Most ML research is focused on the Drug discovery part, since there is a larger quantity of data available that is more convenient to input into a computer.
Background bioactivity / ADMET modeling
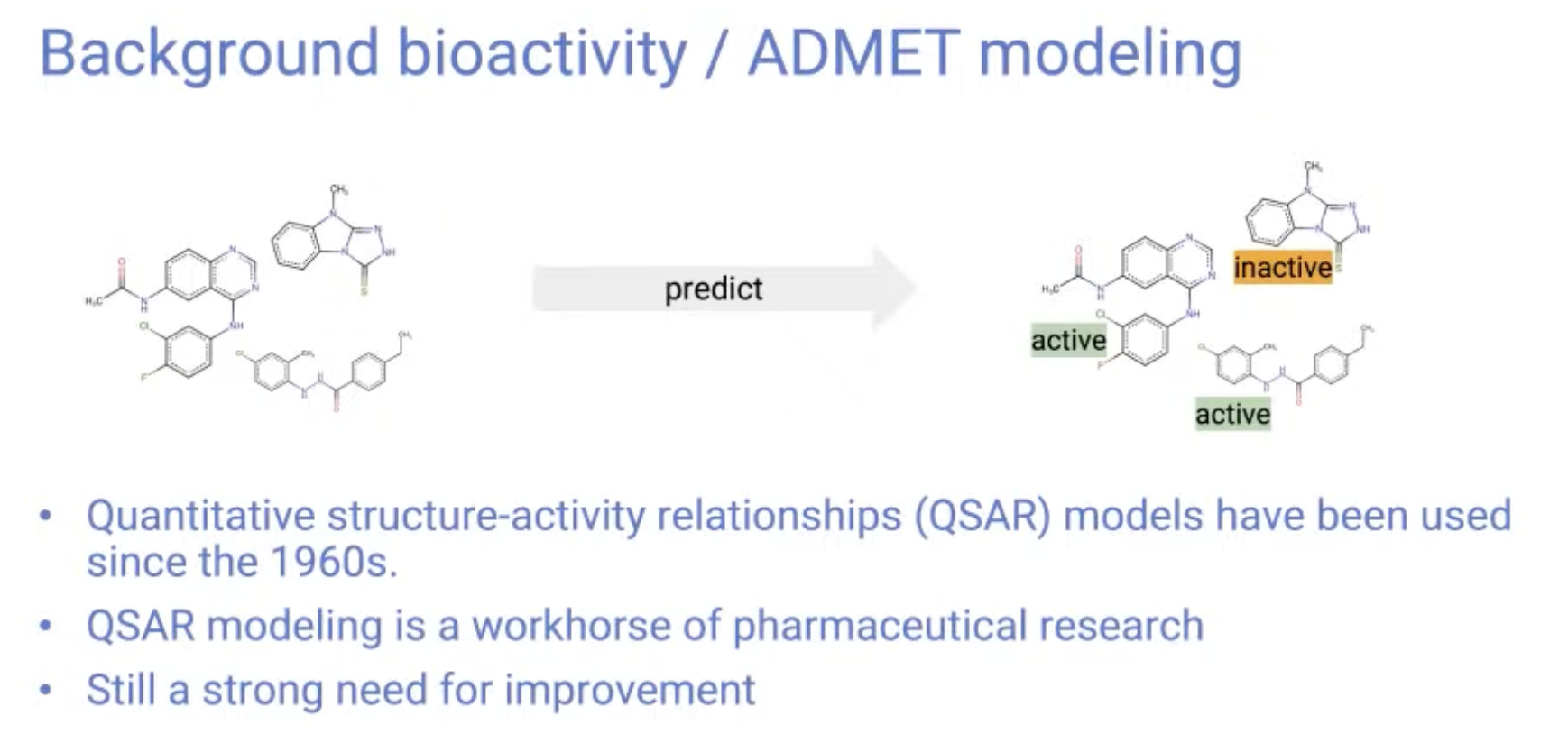
-
Bioactivity modeling have been used since the 1960s.
-
[Research article] Modeling Physico-Chemical ADMET Endpoints with Multitask Graph Convolutional Networks (Molecules, 2019)
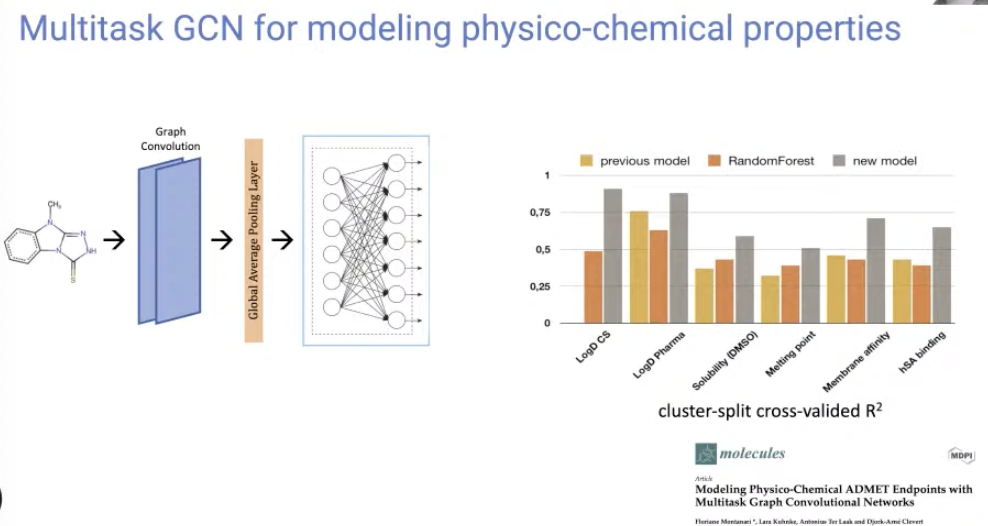
- Multitask GCN for modeling physico-chemical properties.
- Performed a molecular property prediction with GCN in multi-task setting.
- This is an early work in this field using GNNs to predict properties.
-
[Research article] Improving Molecular GCNs Explainability with Orthonormality and Sparsity (ICML, 2021)
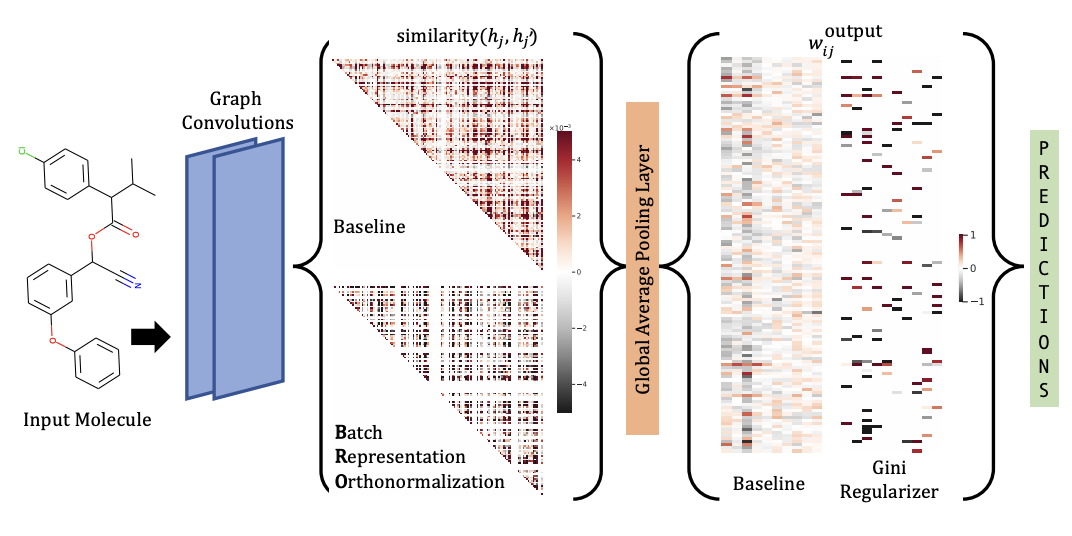
-
Proposed two regularization techniques to improve the accuracy and explainability.
-
Batch Representation Orthonormalization (BRO)
→ encourages graph convolution operations to generate orthonormal node embeddings.
-
Gini regularization
→ applied to the weights of the output layer and constrains the number of dimensions the model can use to make predictions.
-
-
Explainability results
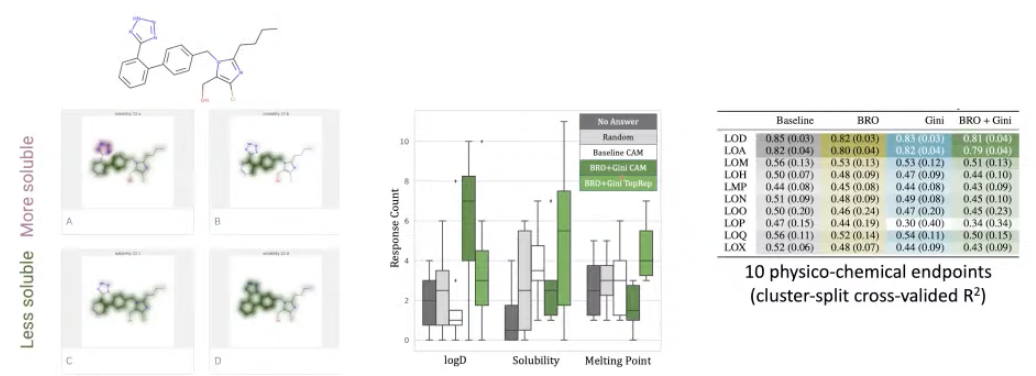
-
-
[Research article] Representation Learning on Biomolecular Structures using Equivariant Graph Attention (LoG, 2022)
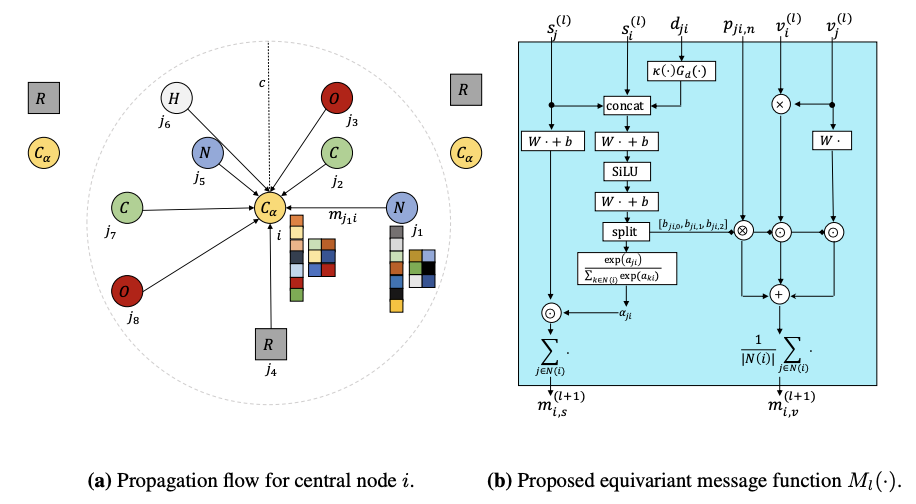
-
Let’s not focus only on invariant feature, but on equivariant feature.
-
EQGAT operates with Cartesian coordinates to incorporate directionality and is implemented with a novel attention mechanism, acting as a content and spatial dependent filter when propagating information between nodes.
-
Performed well on large biomolecule dataset (ATOM3D), and it is efficient.
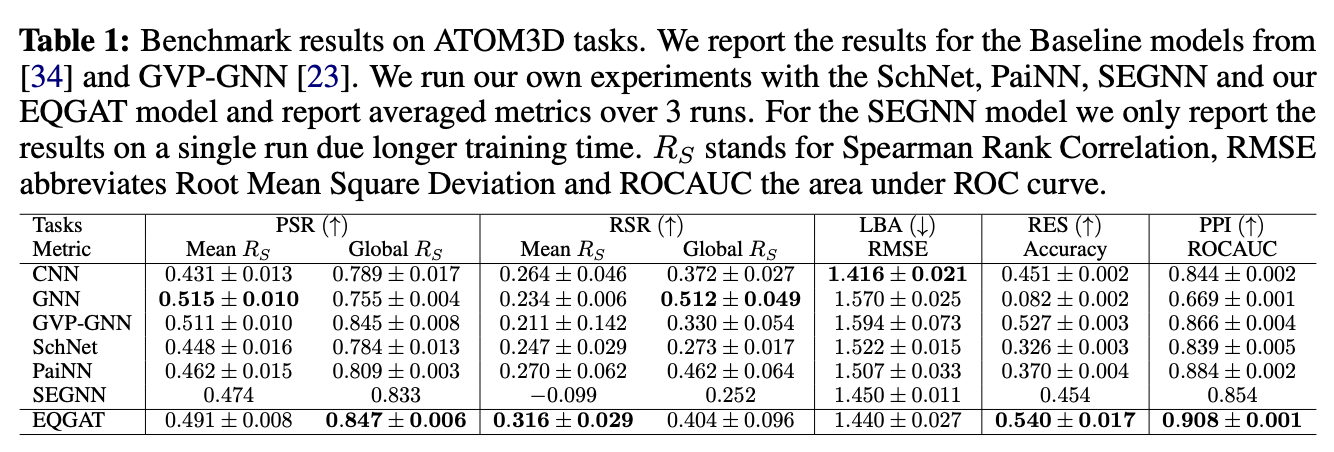
-
The Diversity of Data in Drug Discovery

- There are many types of data in molecule domain, including various spectrometry data, graph, sequence, image, 3D point clouds, …
Molecular Representations for Drug Discovery
-
[Research article] Learning continuous and data-driven molecular descriptors by translating equivalent chemical representations (Chemical Science, 2019)
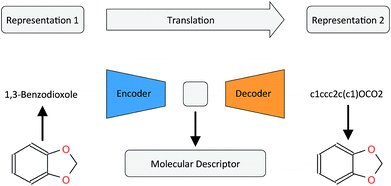
-
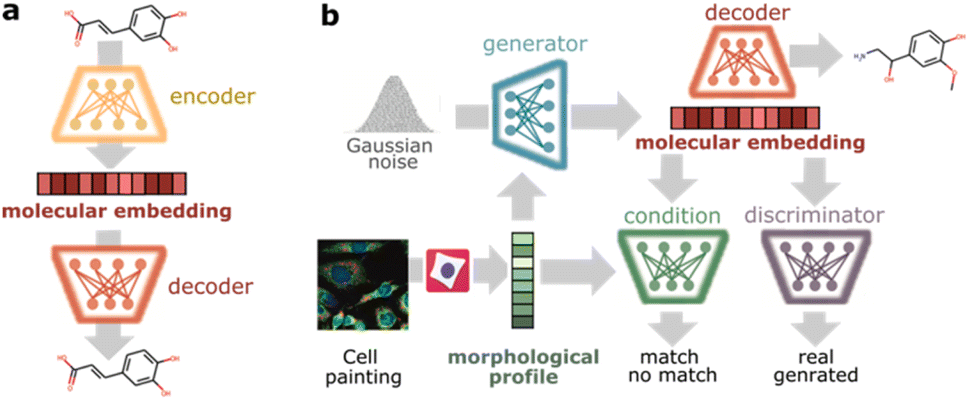
To learn molecule representation, authors made a autoencoder model that translates any SMILES into canonical SMILES. It was good for predicting downstream tasks.
-
Performance results
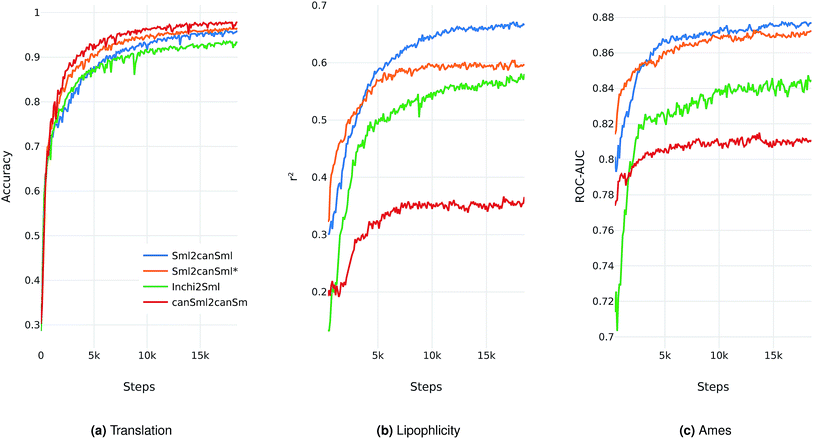
-
-
[Research article] Efficient multi-objective molecular optimization in a continuous latent space (Chemical Science, 2019)
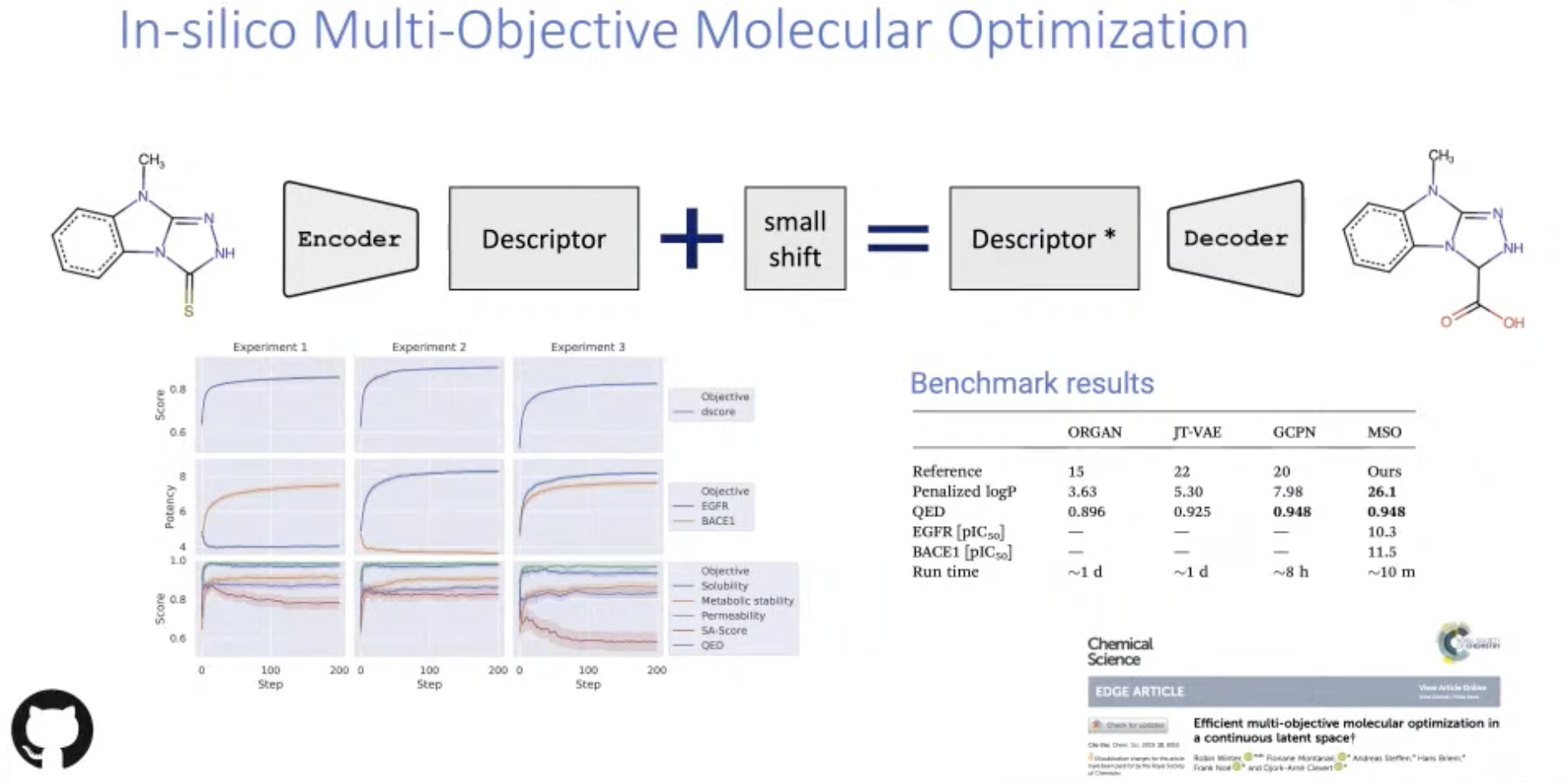
- This method takes a starting compound as input and proposes new molecules with more desirable (predicted) properties.
- Objective function combines multiple in silico prediction models, defined desirability ranges and substructure constraints.
Conditional Molecular de novo Hit Design
-
[Research article] De novo generation of hit-like molecules from gene expression signatures using artificial intelligence (Nature Communications, 2020)
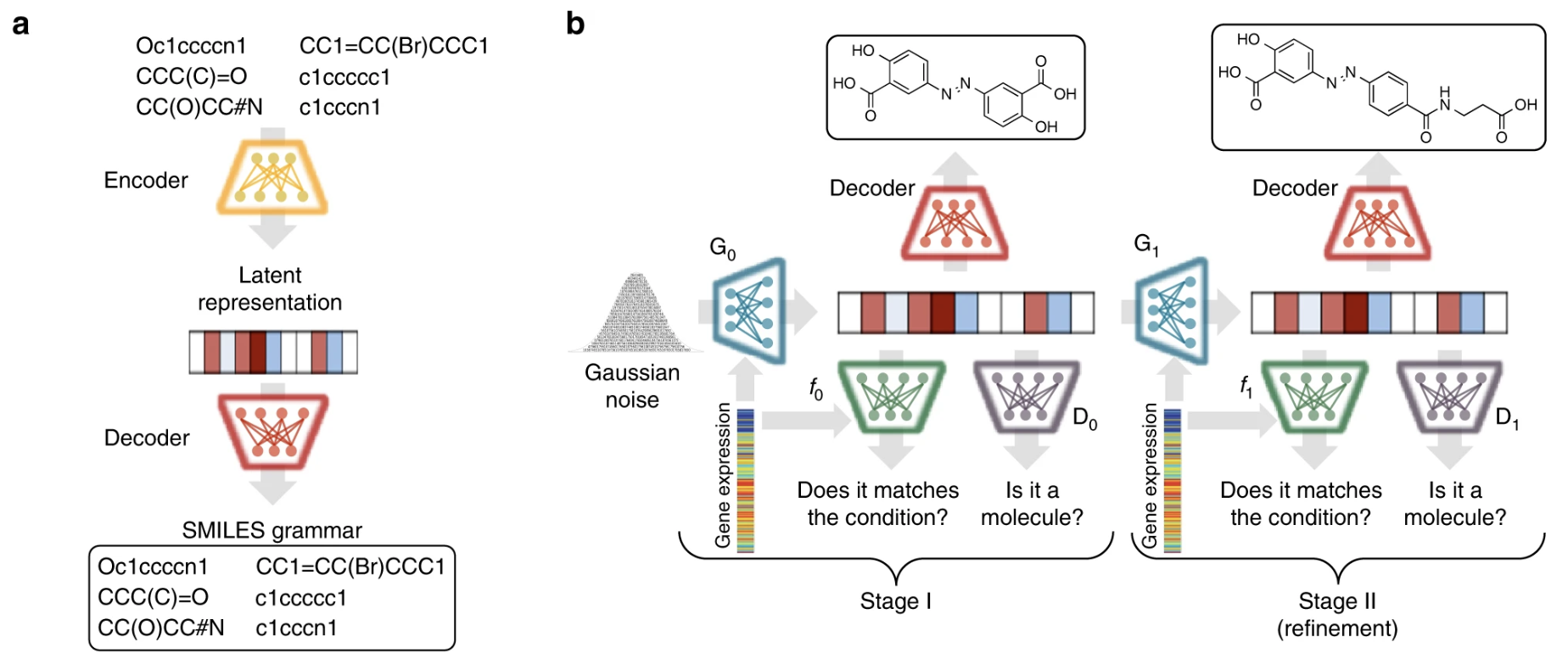
- A generative model (GAN) that bridges systems biology and molecular design, conditioning a generative adversarial network with transcriptomic data.
- When the model is provided with the desired state of gene expression signature, it is able to design active-like molecules for desired targets without any previous target annotation.
-
[Research article] Cell morphology-guided de novo hit design by conditioning GANs on phenotypic image features (Digital Discovery, 2023)
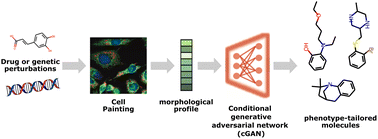
- Utilized cellular morphology (cell painting morphological profiles) to directly guide the de novo design of small molecules. Authors used conditional GAN as a generative model.
Inverse Molecular problems
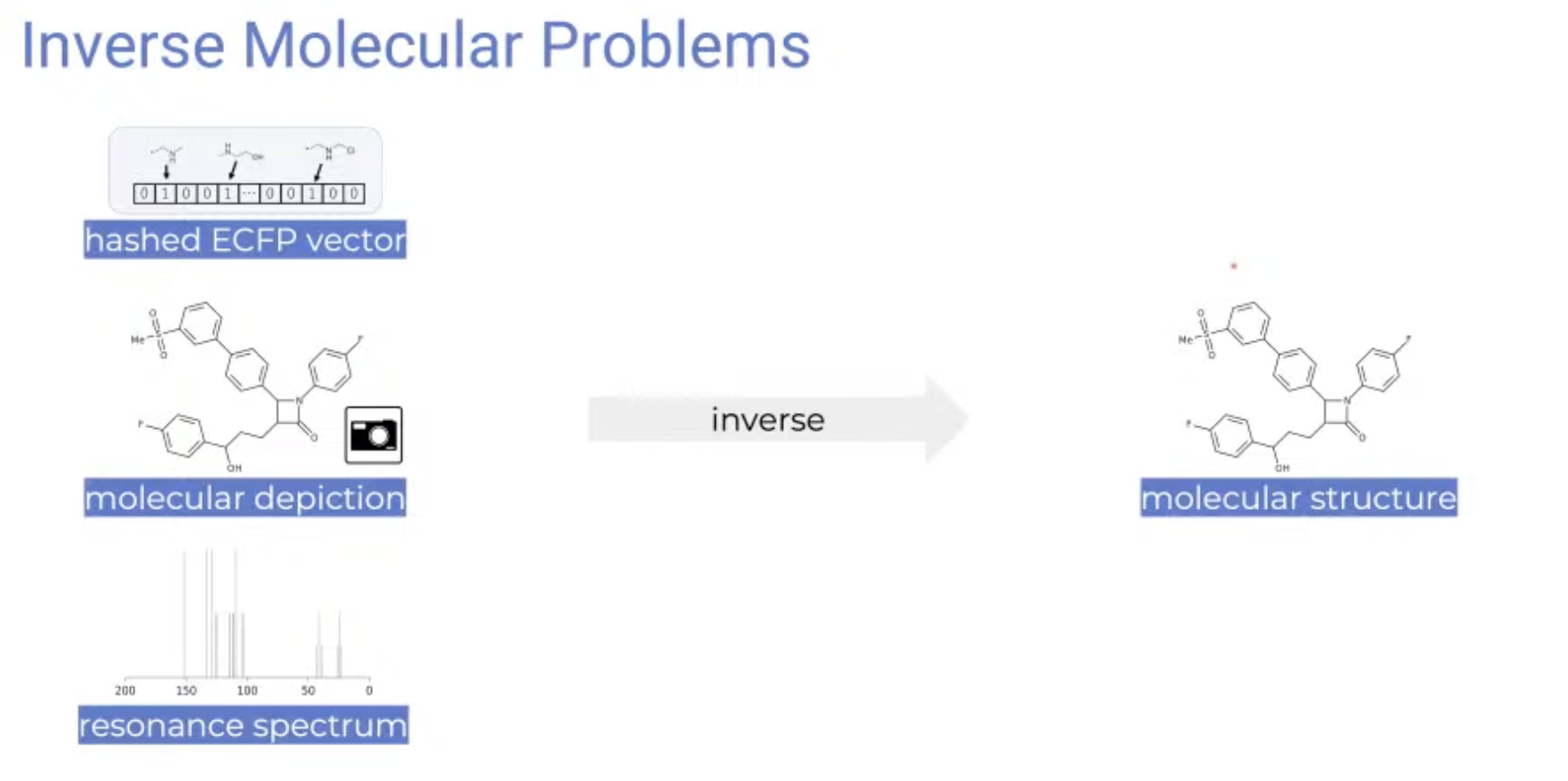
-
Is it possible to inverse fingerprint, molecular depiction, or resonance spectrum into a molecule structure?
-
[Research article] Neuraldecipher – reverse-engineering extended-connectivity fingerprints (ECFPs) to their molecular structures (Chemical Science, 2020)
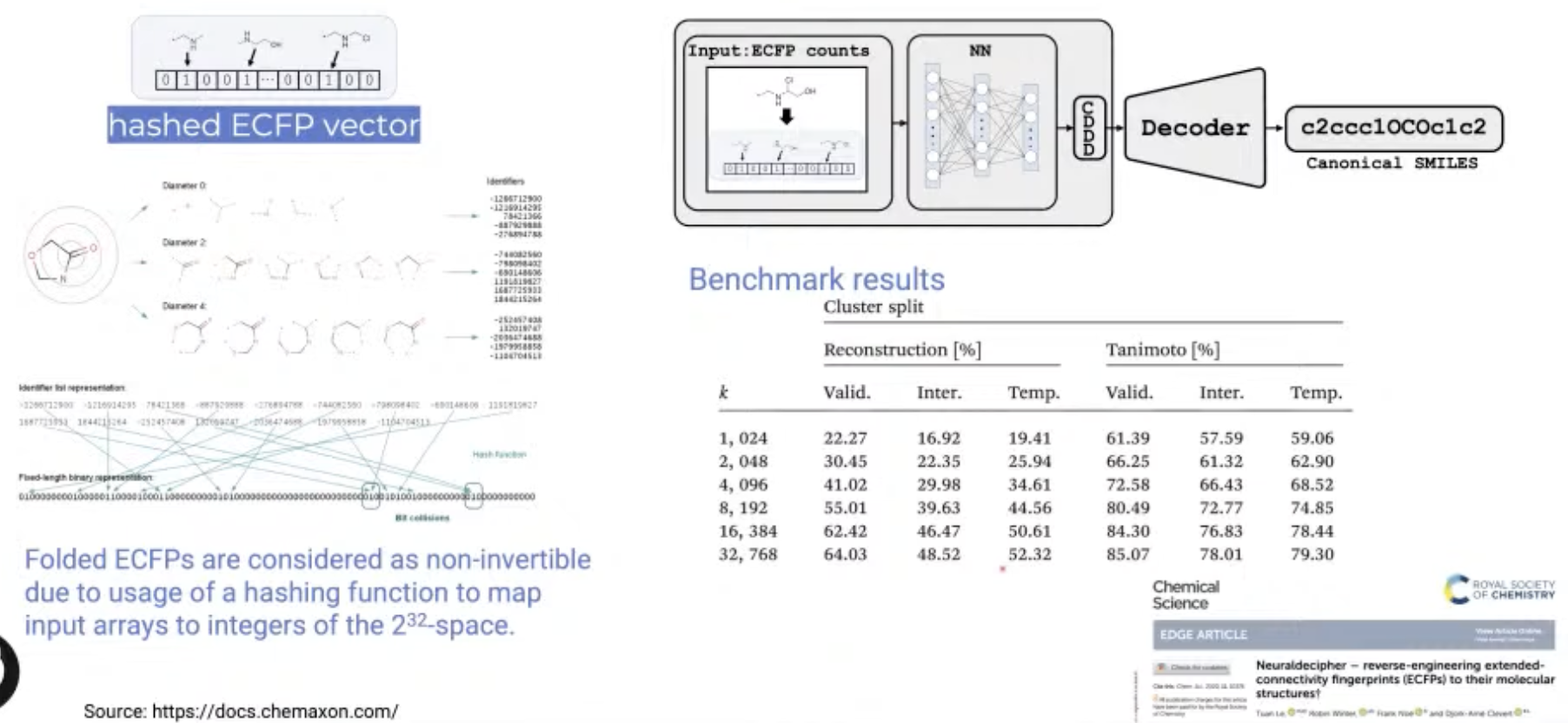
- Since ECFP representation is made with hash function, they are often non-invertible.
- Neuraldecipher is a neural net model that predicts a compact vector representation of compounds, given ECFPs.
- Then utilize another pre-trained model to retrieve the molecular structure as SMILES representation.
- This model were able to correctly deduce up to 69% of molecular structures.
-
[Research article] Img2Mol – accurate SMILES recognition from molecular graphical depictions (Chemical Science, 2021)
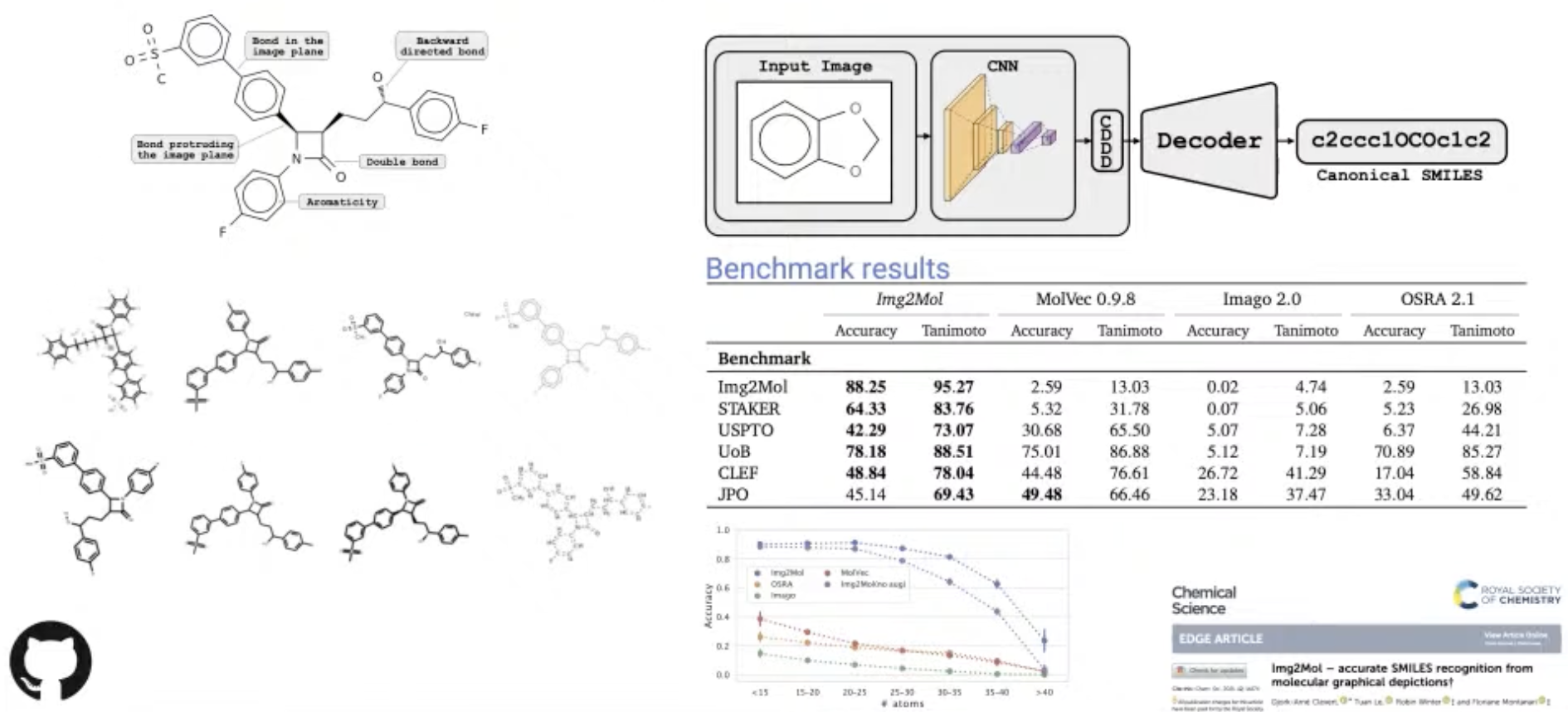
- This model use CNN for molecule depictions and a pre-trained decoder that translates the latent representation into the SMILES representation of the molecules.
- Img2Mol was able to correctly translate up to 88% of the molecular depictions into SMILES.
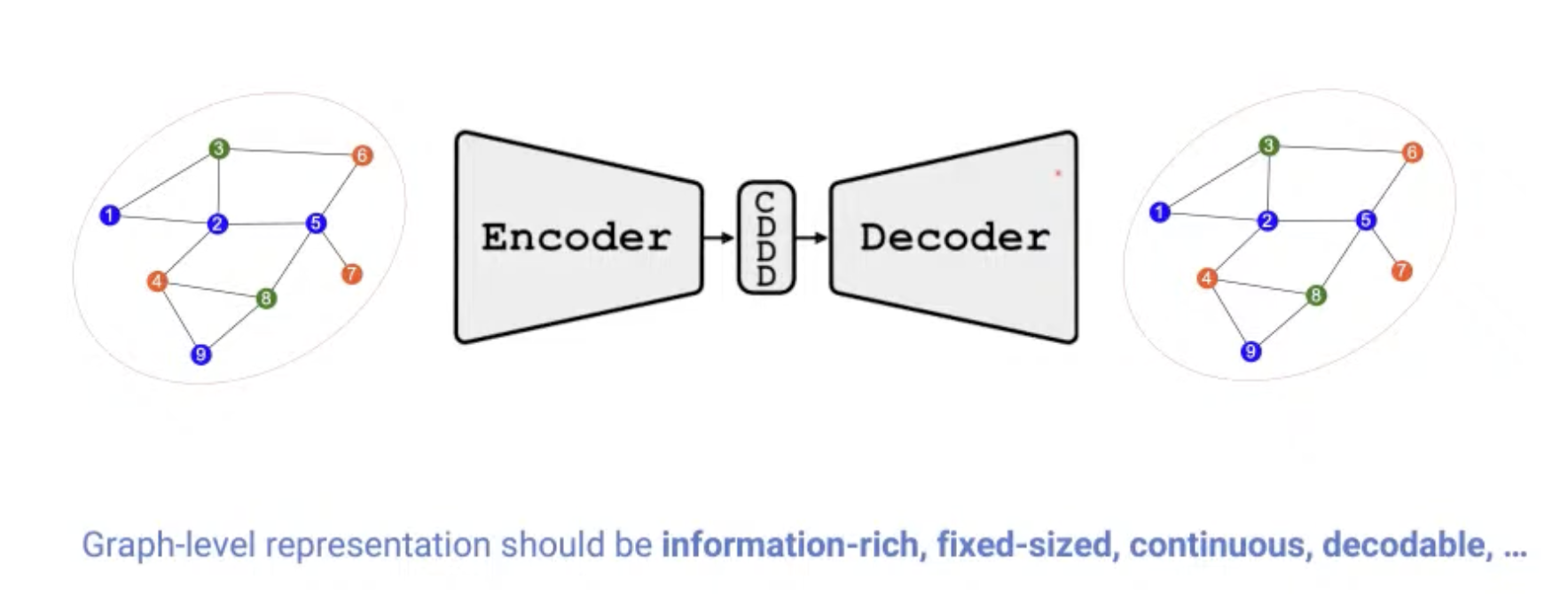
Learning Graph level representation
-
[Research article] Permutation-Invariant Variational Autoencoder for Graph-Level Representation Learning (NeurIPS, 2021, Poster)
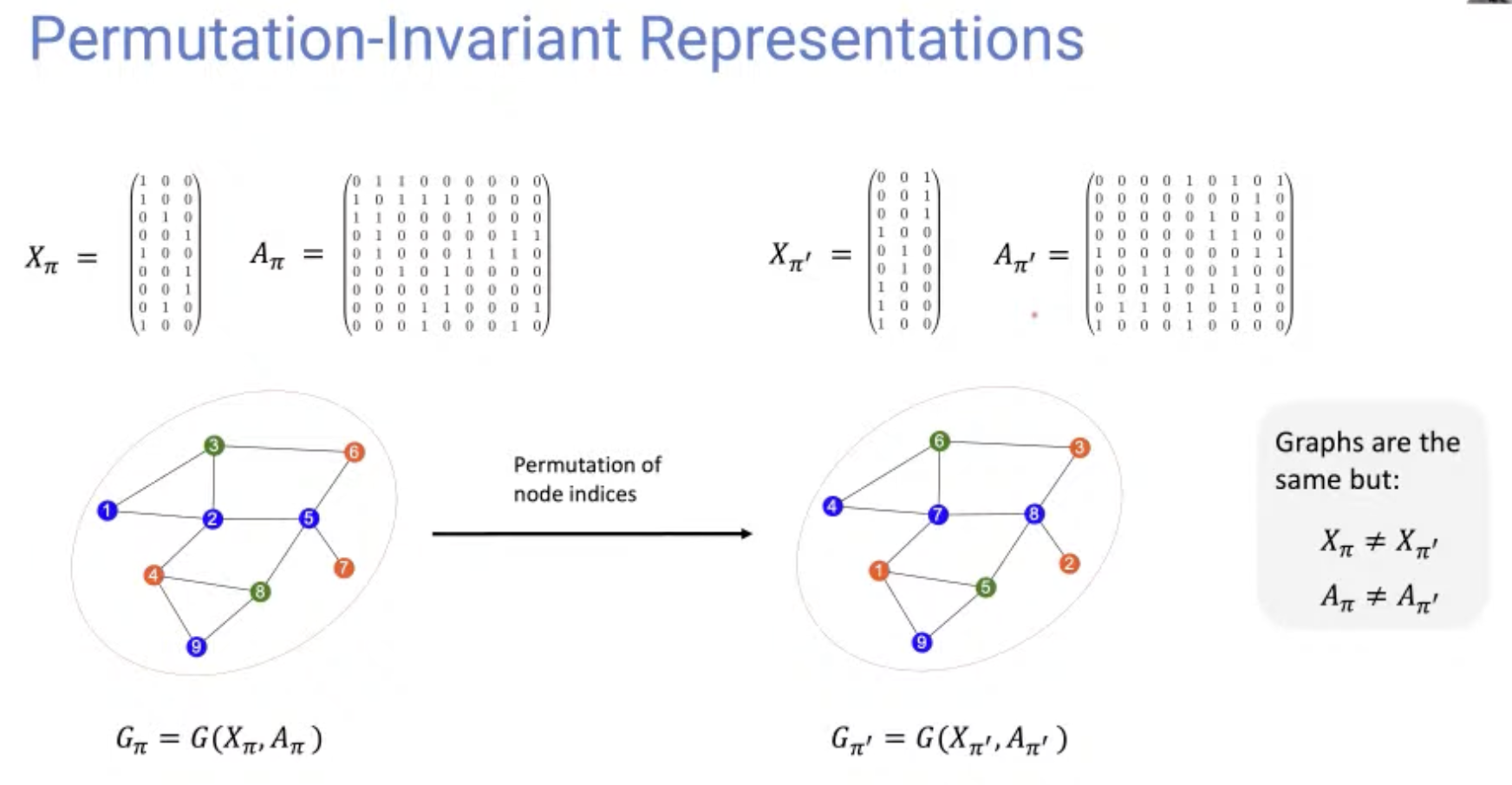
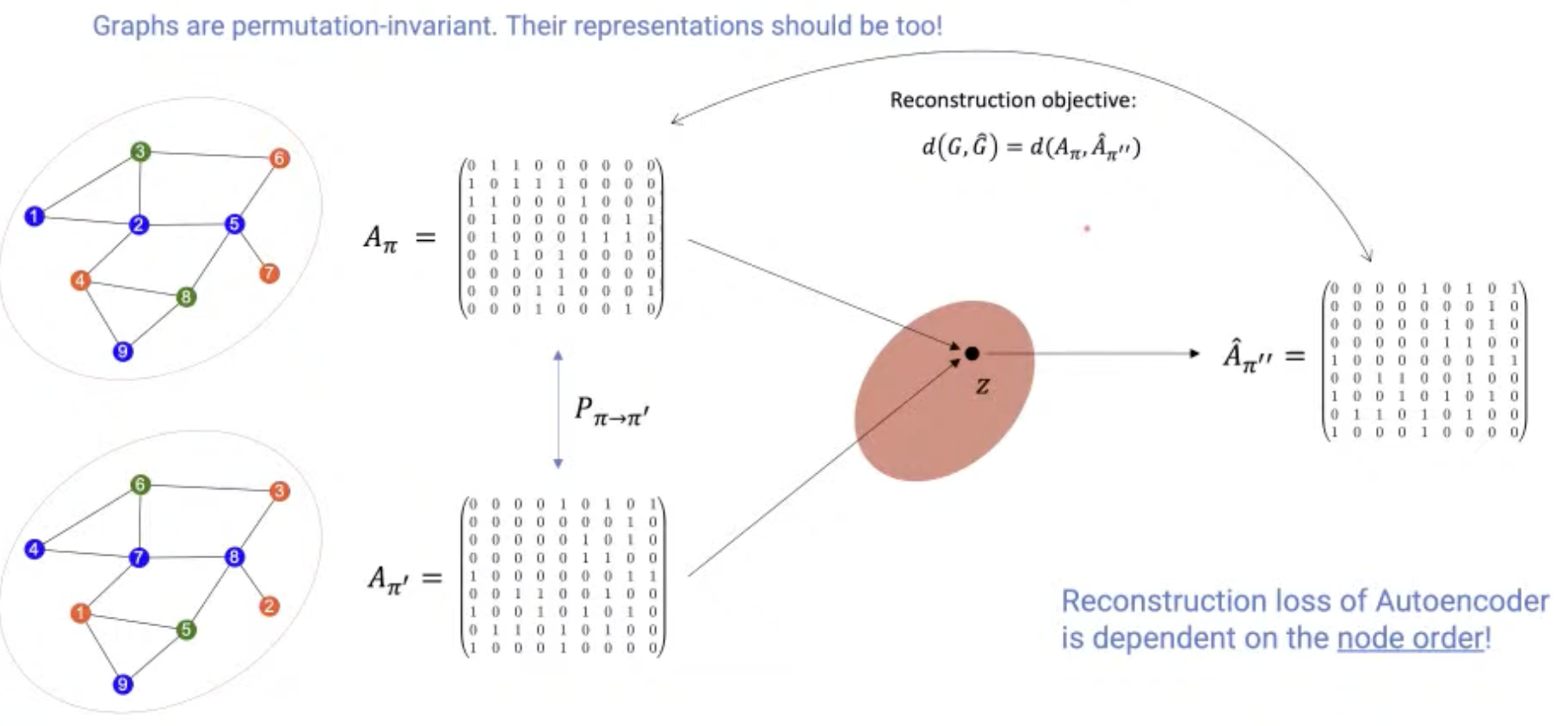
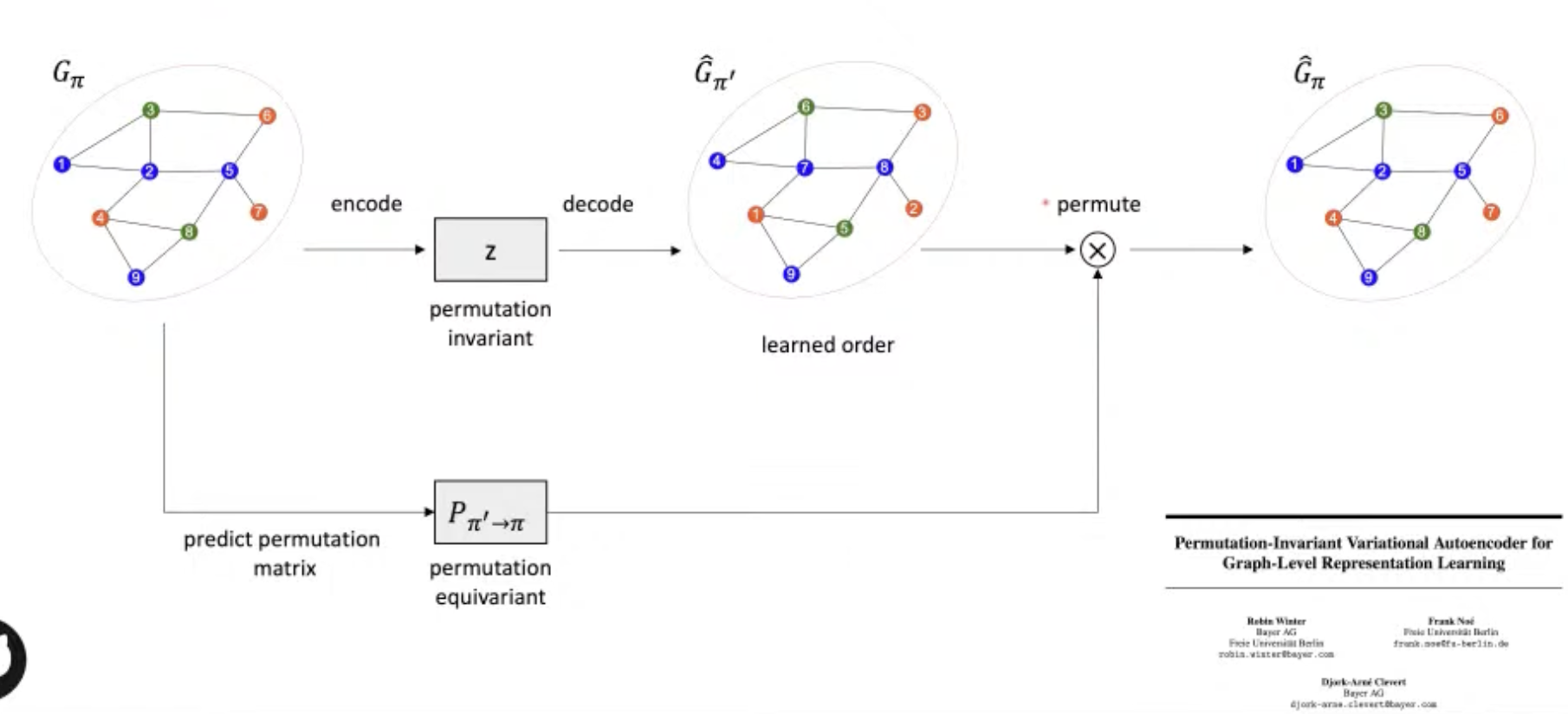
- Graph representation is highly complexed, which can be represented by $(\#\text{ nodes})!$ equivalent adjacency matrices.
- This model indirectly learns to match the node ordering of input and output graph, without imposing a particular node ordering or performing expensive graph matching.
- Showed promising results in graph classification, generation, clustering, interpolation.
Personal Opinion
- It was interesting to see what kinds of research is being performed in big pharmas.
- Big pharmas seem to be more interested in applying ML methods on small problems than creating state-of-the-arts techniques.
- Inverse molecular modeling seems interesting for me.